
Noncompliance in clinical research can pose significant risks to the integrity of a study and the safety of its participants. Whether minor, continuing, or serious, noncompliance requires prompt and effective action to ensure adherence to laws, regulations, and institutional policies. This blog post explores the principles of Corrective and Preventative Action (CAPA) plans, offering best practices for creating and implementing CAPA plans to mitigate noncompliance and safeguard research integrity.
What Is Noncompliance?
Noncompliance is a failure to follow the applicable laws, regulations, or institutional policies governing the protection of participants in research or the requirements or determinations of the Institutional Review Board (IRB).[1]
As defined by the National Institutes of Health (NIH) Intramural Research Program (IRP) Human Research Protection Program (HRPP) Policy Glossary, noncompliance may be minor, continuing, or serious:
- “Continuing noncompliance – A pattern of recurring noncompliance that either has resulted or, if continued, may result in harm to subjects or otherwise materially compromise the rights, welfare, and/or safety of subjects, affect the scientific integrity of the study, or validity of the results. The pattern may comprise repetition of the same noncompliant action(s) or different noncompliant events. Such noncompliance may be unintentional (e.g., due to lack of understanding, knowledge, or commitment) or intentional (e.g., due to deliberate choice to ignore or compromise the requirements of any applicable regulation, organizational policy, or determination of the IRB).
- Serious noncompliance – Noncompliance, whether intentional or not, that results in harm or otherwise materially compromises the rights, welfare, and/or safety of the subject. Noncompliance that materially affects the scientific integrity or validity of the research may be considered serious noncompliance, even if it does not result in direct harm to research subjects.”
Noncompliance with the protocol, Standard Operating Procedures (SOPs), Good Clinical Practice (GCP), and/or applicable regulatory requirements by an investigator, institution, sponsor, or their staff should be acted upon promptly to secure compliance. If noncompliance significantly affects or has the potential to affect participants’ rights or welfare, a root cause analysis should be performed and a Corrective and Preventative Action Plan (CAPA) implemented. CAPA is crucial in identifying and addressing the root causes of noncompliance, ensuring that corrective measures are effective and preventing future occurrences of noncompliance.[2]
What Is a CAPA?
A CAPA consists of a few key elements:[3]
- A Correction is an action to eliminate a detected nonconformity (nonfulfillment of a requirement).
- Corrective Action is an action to eliminate the known cause of a nonconformity or other undesirable situation and prevent recurrence.
- Preventative Action is an action to eliminate the potential cause of a future nonconformity or other undesirable situation and prevent occurrence.
The purpose of corrective and preventative action is to collect and analyze information, identify and investigate problems, and take appropriate and effective corrective and/or preventative action to prevent their occurrence or recurrence.[4]
How Is a CAPA Created?
In establishing and maintaining a CAPA, depending on the size and complexity of your research location(s), there should be procedures in place to:[5]
- Analyze
- Investigate
- Identify
- Verify/Validate
- Implement
- Disseminate
- Review
- Record
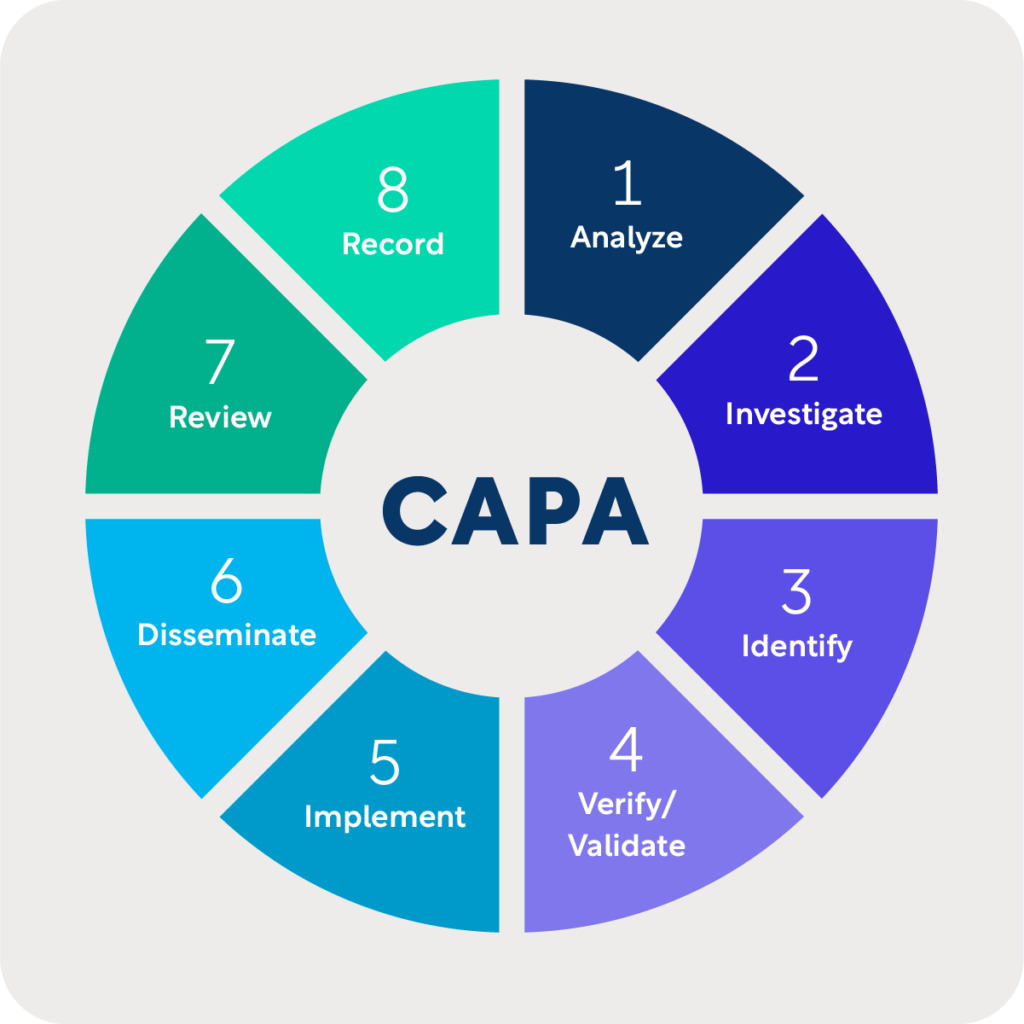
In analyzing, you should investigate internal processes, work operations, audit reports, records or source documents, complaints, and other sources of monitoring to identify existing causes of nonconformity.
Future potential causes of nonconformity should also be considered. Identify critical data and processes that if inaccurate, not performed, or performed incorrectly, may lead to concerns for participants’ rights, welfare, or safety. Common items or areas identified as critical are:[6]
- Confirmation that informed consent was obtained properly.
- Adherence to protocol eligibility criteria designed to exclude individuals for whom the investigational product may not be safe or for whom the product is otherwise inappropriate.
- Procedures to correctly administer and/or dose the investigational product.
- Conduct and documentation of procedures and assessments related to study endpoints, protocol-required safety assessments, evaluating/documenting/reporting serious or unanticipated adverse events.
Once sources of reportable actions have been identified, launch an investigation into the root cause. A commonly cited root cause of noncompliance is current or past employee’s behavior or actions. While human error does occur, problems that appear to stem from individuals may be the result of a flaw in the systems or processes. When flaws are left unidentified or unchanged, the problem will reoccur.
Identify what action(s) are needed. This could be:
- A determination that no further action is necessary.
- Correction.
- Corrective action.
- Preventative action.
When prioritizing actions, consider a risk-based approach. Risk per ISO 14971 is defined as the “combination of the probability of occurrence of harm and severity of that harm.” The Food and Drug Administration (FDA) agrees that the “degree of corrective and preventative action taken to eliminate or minimize actual or potential nonconformities must be appropriate to the magnitude of the problem and commensurate with the risk encountered.”[7] You would use a risk-based approach to rank the nonconformities from major to minor impact and proceed from high to low risk until all areas are addressed.
Common actions include:
- Additional training of investigators and staff. All employees should receive study-specific training that includes discussions of the trial design, protocol requirements, study monitoring plan, applicable SOPs, appropriate monitoring techniques, and electronic systems.[2]
- Clarification of the protocol requirements.
- Protocol amendments or protocol clarification letters.
- Creating a Note to File (NTF) to explain and document nonconformities in the research record.
- Updates to SOPs.
- Updates to electronic or manual record keeping systems or source documentation.
A CAPA may also include a process for verifying or validating that corrective or preventative actions are effective. This would include collecting data on the corrective action you have implemented.[8] If the results indicate further problems, additional corrective measures or deeper root cause analysis may be required.
When implementing the solutions:
- Record changes that were needed to correct and prevent the identified problems.[9]
- Disseminate the plan to those responsible for assuring the quality of such product or the prevention of such problems.
- Provide information to management, as appropriate, on the identified issues as well as any corrective or preventative actions taken.
- Ensure the issues, corrective and preventative actions, and all relevant activities are documented in the research records.
Sample CAPA Template:
| Sponsor Protocol #: | IRB Protocol #: |
| PI Name: | IRB Study #: |
| Protocol Title: |
| Date Issue(s) Occurred |
| Outside of the IRB, to Whom Has This Issue Been Reported? |
| Description of the Problem(s): Provide a description of the issue with enough information that someone not directly involved will be able to understand the scenario. |
| Source of the Problem(s): What is the root cause of the issue(s)? |
| Were Any Participants Impacted? If so, Describe: |
| Correction: What immediate actions were taken to eliminate risk of harm to participants’ rights, safety, or welfare (if applicable): Date: When was this action taken? |
| Corrective Action: What actions will be taken to eliminate the known cause of the existing issues and prevent reoccurrence? |
| Preventative Action: What actions will be taken to eliminate potential future issues and prevent reoccurrence? |
| Documentation: How will these actions be documented and conveyed to stakeholders? |
| Effectiveness Check: Describe what actions will be taken to verify the effectiveness of your corrective and preventative actions. Describe who will be responsible for completing the evaluation. Provide a timeline for your effectiveness check. |
| Person Completing This Form: Date: |
References
- NIH (National Institutes of Health) IRP (Intramural Research Program) Human Research Protection Program (HRPP) Policy Glossary
- E6(R2) Good Clinical Practice: Integrated Addendum to ICH E6 (R1)–section 5.20
- ISO 9000:2005
- Corrective and Preventive Actions (CAPA) https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-guides/corrective-and-preventive-actions-capa [1] 21 CFR 820.100
- 21 CFR 820.100
- FDA: Guidance for Industry: Oversight of Clinical Investigations – A Risk-Based Approach to Monitoring
- 61 Fed. Reg. at 52633-52634, Comment 159
- ISO 9001
- 21 CFR 820.100
Don't trust your study to just anyone.
WCG's IRB experts are standing by to handle your study with the utmost urgency and care. Contact us today to find out the WCG difference!