
With the COVID-19 pandemic, many physicians were for the first time faced with individual patient expanded access as a possible course of action. COVID-19 is not the only disease that exists for which expanded access may be a good option. Whether for treatment of COVID-19 or not, expanded access can appear to be a daunting endeavor, especially for those physicians not part of an institution or without an established relationship with an institutional review board.
This paper will help provide guidance on starting the expanded access request process for those physicians who want to provide an expanded access option to their patient as quickly as possible.
There is more than one kind of expanded access1, as outlined by the Food and Drug Administration (FDA), and this paper only addresses individual patient expanded access; that is, a treatment plan for one person for an unapproved product. Other instances of expanded access, such as an expanded access program for multiple people, are outside the scope of this paper.
Programs for multiple people (intermediate size or large populations) are in many ways treated more like a research protocol. When the investigational therapy is a gene therapy/ gene transfer therapy, some Expanded Access protocols require approval by an Institutional Biosafety Committee (IBC) under the NIH Guidelines for Research Involving Recombinant and Synthetic Nucleic Acid Molecules (NIH Guidelines)2, as well as by an IRB.
In 2019 the NIH Guidelines were amended to remove the requirement for IBC approval specifically for Individual Expanded Access treatment plans, so IBC approval is not required for the types of submissions covered in this paper.
Starting the Expanded Access Request Process
The first step in the process is ensuring that the patient is open to treatment with an investigational product, and determining that there is an appropriate investigational product for the condition or disease to be treated. The next step is contacting the company/manufacturer of that product to confirm their agreement to provide the investigational product for this patient, which will likely involve agreement that the physician will follow all necessary regulatory procedures and will report safety issues back to the manufacturer. After that, FDA approval is required (see Figure 1).
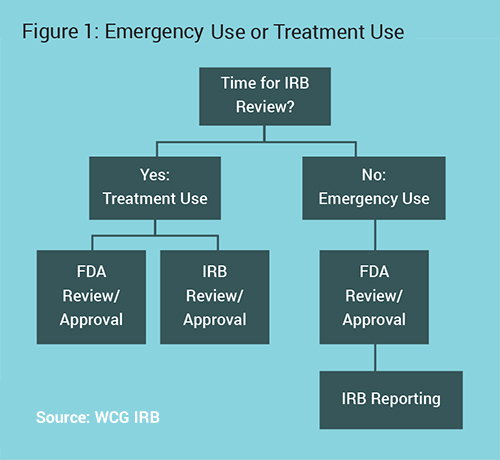
The main decision point is if there is sufficient time for prospective Institutional Review Board (IRB) review or not. Physicians should talk to their IRB to determine an expected timeline, as IRBs often prioritize these reviews. They may be able to review much quicker than a standard research protocol. The subsequent processes change based on this point.3
Emergency use of experimental devices does not require prospective FDA or IRB review if a treating physician has access to an investigational device, determines it is the best course of treatment for a patient with a life-threatening condition, and has no time to obtain FDA approval.
The physician should follow as many patient protection procedures as possible, including:
- Patient/Legally Authorized Representative (LAR) consent
- Institutional clearance
- IRB chair concurrence
- Independent physician review and agreement
- Device manufacturer authorization
- IRB and FDA reporting after use is required.4
Emergency Use
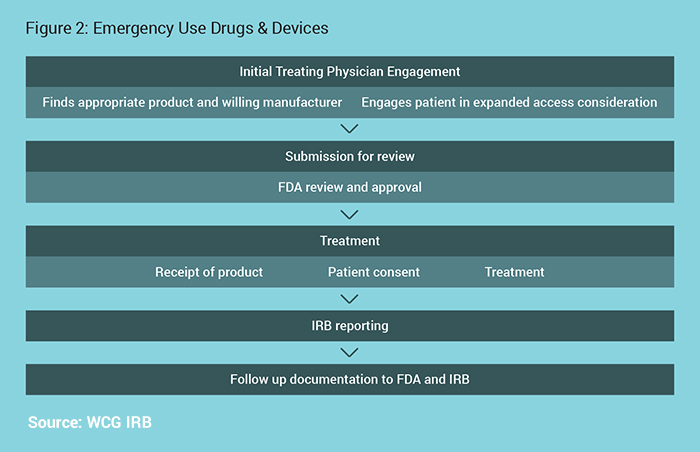
The Emergency Use process is outlined in Figure 2.
For Devices:
If all of the criteria are met, an unapproved device may be used in an emergency situation without prior approval by the FDA if needed. If there is time, FDA has emergency phone numbers to grant immediate review of the proposed use.
For Drugs:
Either call or use another means of rapid communication, or submit form FDA form 3926. Unlike research, the FDA does not expect that a protocol document would be required. For example, information from a patient history and treatment plan on the form 3926 is sufficient.5
For Drugs & Devices:
Prospective IRB review is not required. Reporting to the IRB within 5 days of use is required.6
Treatment Use
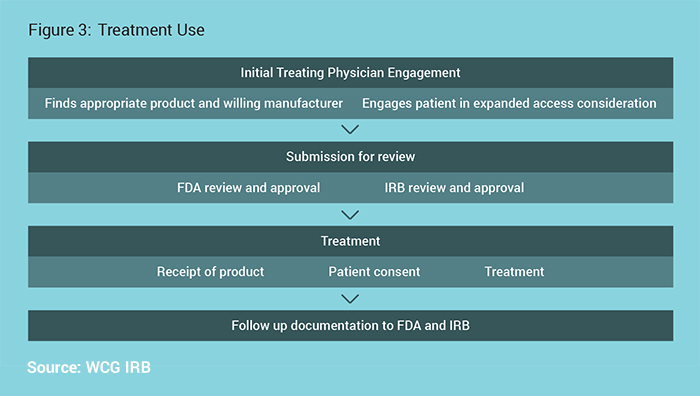
The Treatment Use process is outlined in Figure 3.
FDA will authorize use with prospective IRB review required if there is sufficient time for prospective IRB review. Their authorization will clearly note if IRB review is required. Of note, IRB review is never required prior to FDA review, and the IRB and FDA review can occur in parallel. For those physicians who are part of an institution or otherwise obligated to one IRB, contact the IRB and follow the IRB’s procedures. If under no obligation to another IRB, an independent IRB may be used.
For Drugs:
While the use of an investigational product would otherwise need to be reviewed at a convened meeting, physicians submitting an individual patient expanded access Investigational New Drug (IND) using FDA Form 3926 may select the appropriate box (10b) on that form to request that FDA grant a waiver from full IRB review. Waiving full IRB review often saves significant time, and does not diminish the protections for the patient. FDA recommends IRBs establish specific procedures for a single IRB member to review these submissions, should the treating physician request a waiver from review by the convened IRB. According to their guidance on the topic, FDA concludes that such a waiver is appropriate for individual patient expanded access INDs when the physician obtains concurrence by the IRB Chairperson or another designated IRB member before treatment use begins. If not using FDA Form 3926 and using FDA Form 1571 instead, a physician submitting an individual patient expanded access IND may include a separate waiver request.7
For Devices:
The request may be submitted directly by the physician to the FDA CDRH address with specific information, such as the specifics of the case and why the investigational device is the best choice. Noting the patient protection measures include the concurrence of the IRB chairperson allows for that concurrence to be submitted for IRB review.8
For their review, the IRB should have a dedicated submission form for these requests to reduce the confusion between a standard research review and an individual patient expanded access, and to collect only the necessary information relevant to this type of submission, limiting the information required and thus the time to complete it.9
Informed Consent in Expanded Access
Because of the difference in clinical consent and consent under the regulations regarding the use of an investigational product, the IRB review may appear to focus their review primarily on the consent form. The primary purpose of IRB review is to assure that the rights and welfare of human subjects are protected. In the case of individual patient expanded access, where the treatment of the individual is inherently for the benefit of the individual’s welfare, the rights of the person to make an informed choice are paramount. The IRB’s role in this is to determine that informed consent is obtained in accordance with and to the extent required by Federal requirements under the regulations.10
Emergency Use:
The consent issues for emergency use are complex. Emergency use of a device may not allow time for informed consent (for example, if a decision needs to be made during an operative procedure), and IRB review of the consent form may occur after the device has been implemented or after the unapproved drug/biologic has already been given to the patient. Nevertheless, the use of investigational products requires written informed consent11 as described in the regulations.12
The most recent FDA guidance on individual patient expanded access requests during the COVID-19 provided several examples on how consent and documentation may
be obtained when physical distancing is required. Examples included procedures with transmitting a photograph of the signed document or a witness to the consent process and documentation.13 The IRB review of the consent process should account for this potential.
Individual patient expanded access requires documentation of informed consent with a consent form, as noted above. The requirements for the consent form for expanded access are no different than those for research; however, they may be significantly more than someone unfamiliar with a research consent form might expect. The product manufacturer should be able to provide a consent form from a research study if nothing more than for the most recent risk information. If that is used, any changes to match the individual person’s treatment plan need to be made, for example, in the procedures and contraception sections. If the IRB has a template form, using that template for non-specific sections such confidentiality and questions will also decrease the chance of needing edits.
IRB Review of Consent in Treatment Use vs. Emergency Use:
For treatment use, the IRB’s prospective review of the consent form and consent process will follow their usual process for review. As noted, since the review is not the same as review by the convened committee for research, there will be differences, such as no IRB approval stamp on the consent form.
For emergency use, which is authorized to proceed prior to IRB review, the consent process and form is sent to the IRB after the fact. However, as the requirements for consent are not different, the IRB may require the person be re-consented with an updated consent form if their form was not used for creation.
Summary
The unfamiliar situation of an individual patient expanded access can seem confusing or complicated. By following these steps of securing access to the investigational product, obtaining any necessary FDA documentation, and creating a consent form made using with the drug manufacturer’s and IRB’s templates, an individual patient expanded access will proceed more smoothly and quickly.
Physicians applying for Expanded Access may also find it useful to use the eRequest website/app created by the Reagan Udall Foundation, which provides a step-by-step guide to the submission process.
References
- U.S. Department of Health and Human Services, Food and Drug Administration. “Expanded Access | Information for Physicians.” Accessed 18 November 2021. https://www.fda.gov/news-events/expanded-access/expanded-access-information-physicians
- U.S. Department of Health and Human Services, National Institutes of Health, Office of Science Policy. “NIH Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules.” Accessed 12 November 2021. https://osp.od.nih.gov/wp-content/uploads/2019_ NIH_Guidelines.htm
- U.S. Department of Health and Human Services, Food and Drug Administration. “Expanded Access Program Report.” Published May 2018. Accessed 12 September 2021.
- U.S. Department of Health and Human Services, Food and Drug Administration. “Expanded Access for Medical Devices.” Accessed 12 November 2021. https://www.fda.gov/medical-devices/investigational-device-exemption-ide/expanded-access-medical-devices
- U.S. Department of Health and Human Services, Food and Drug Administration. “Emergency use of an Investigational Drug or Biologic: Guidance for Guidance for Institutional Review Boards and Clinical Investigators.” Published January 1998. Accessed 12 November 2021. https://www.fda. gov/regulatory-information/search-fda-guidance-documents/emergency-use-investigational-drug-or-biologic
- Code of Federal Regulations, Title 21, Food and Drugs, Department of Health and Human Services. Part 56, Institutional Review Boards, Subpart A, General Provisions, Section 104, Exemptions from IRB requirement
- U.S. Department of Health and Human Services, Food and Drug Administration. “Emergency use of an Investigational Drug or Biologic: Guidance for Guidance for Institutional Review Boards and Clinical Investigators.” Published January 1998. Accessed 12 November 2021. https://www.fda. gov/regulatory-information/search-fda-guidance-documents/emergency-use-investigational-drug-or-biologic
- U.S. Department of Health and Human Services, Food and Drug Administration. “Expanded Access for Medical Devices.” Accessed 12 November 2021. https://www.fda.gov/medical-devices/investigational-device-exemption-ide/expanded-access-medical-devices
- U.S. Department of Health and Human Services, Food and Drug Administration. “Expanded Access to Investigational Drugs for Treatment Use —Questions and Answers. Guidance for
Don't trust your study to just anyone.
WCG's IRB experts are standing by to handle your study with the utmost urgency and care. Contact us today to find out the WCG difference!